
Conesa paper offers guidelines for more precise RNA-seq data analysis
 A team headed by Genetics Institute faculty member Ana Conesa is attempting to bring some order to the Wild West of RNA-seq data analysis.
A team headed by Genetics Institute faculty member Ana Conesa is attempting to bring some order to the Wild West of RNA-seq data analysis.
Although the technology– used to study gene expression– has been available for almost a decade, Conesa, professor of microbiology and cell science, said researchers often take a haphazard approach to using it.
Its reputation as a particularly reliable means of analysis, and higher operational cost, has led some to perform fewer replications than they would with other methods– leading to errors in their results.
“People tend to think RNA-seq is an established methodology, and it’s perfect,” Conesa said. “But it’s not.”
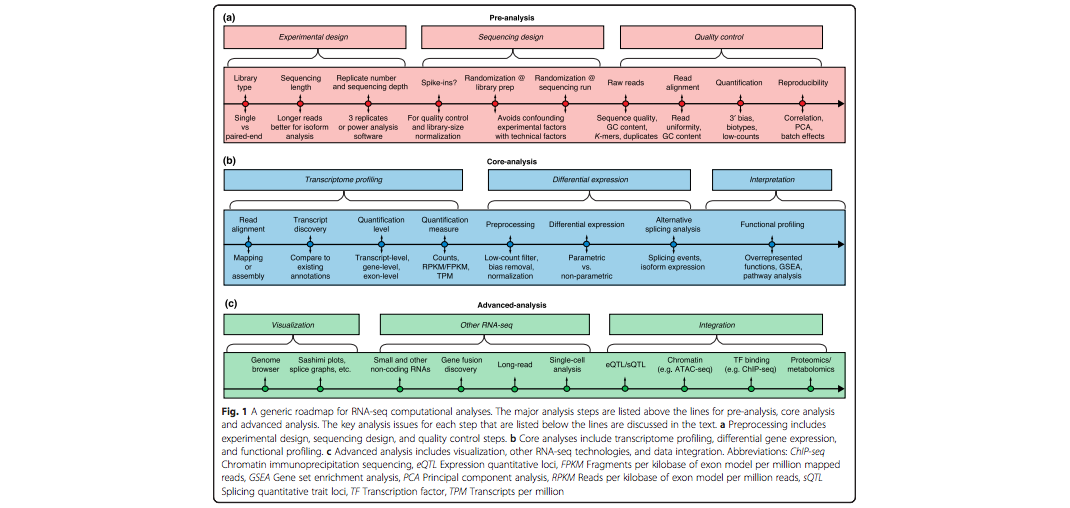
Her paper, “A survey of best practices for RNA-seq data analysis” published in January by Genome Biology establishes some guidelines for using the technology.
Conesa emphasized the importance of careful experimental design, quality control and pre-process analysis.
RNA-seq is often used in combination with other types of methods of analysis that measure additional components such as DNA methylation and chromatin features. In the paper,
Conesa’s team addresses how to more precisely combine multiple
methods of data analysis.
Despite the comparatively higher cost of using RNA-seq, Conesa said replicating the experiment multiple times is essential. The notion that RNA-seq is accurate enough to only necessitate a single analysis is a myth.
“Also, what we made clear here is that RNA-seq can be used in different applications,” Conesa said. “You can use it for different purposes, and each application has a different analysis pipeline.”
However, different applications call for different input parameters and planning.
In the paper, they discuss different pipelines for the different applications.
The publication also includes suggestions for software that can be used in analysis.
“I think people that are not really experience with RNA-seq analysis should read the paper before they go into business,” Conesa said.
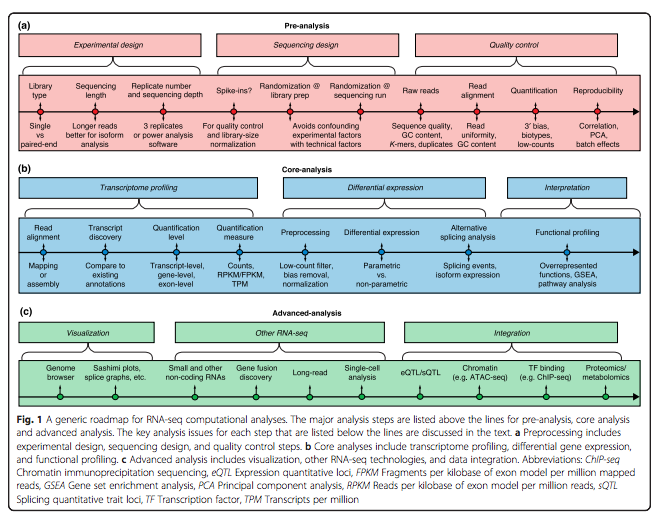
Source: “A survey of best practice for RNA-seq data analysis”